集団遺伝学的解析用ソフトウェア「Arlequin」 の使い方
こんにちは~ ガイです。
Arlequin は多機能なソフトウェアですべては網羅しきれないので、とりあえず自分が使った機能の使い方だけ載せていきます。
具体的には
- Mismatch Distribution 解析
- Fst 解析
- Neutrality tests
です。それ以外の機能についてを求めている場合、以下を読んでもあんまり意味が無いと思うのでそっ閉じしてくださいね。
ちなみにArlequin の公式マニュアルが3.5のしかないのでわたしはそれを見ていろいろ言ってます。
Manual Arlequin ver 2.0 (unibe.ch)
- 「Arlequin 動かない問題」にぶち当たっていませんか?
- 1.インストール
- 2.DNAsp を用いてインプットファイルを作成
- 3.Fst の計算
- 4.Mismatch Distribution 解析
- 5.Neutrality tests
初めに…
「Arlequin 動かない問題」にぶち当たっていませんか?
もしかしたらバグが治っているかもしれませんが、2年前わたしが導入したとき、Alrequin のArlequin 3.5.2.2 は最後までファイルを書き出ししてくれませんでした。昨年度の同期も同じ悩みを抱えていたのでわたしのPC の問題ではないと思います。
解決策としては「Arlequin 3.5.1.2」を使うことがあげられます。
残念なことに今確認したら3.5.1.2 のインターフェースつきのもの、「winarl35.zip」はリンク切れでした。これからご紹介する方法はインターフェースつきのものでの説明になるので、何らかの方法で手に入れてください…先輩に動くバージョンのフォルダをコピーさせてもらうとか…あれ、そういう再配布は違法なのかな?そこまでカバーしきれなくてごめんなさい。でもあなたのソフトウェアが途中までしか動かないのは高確率でバージョン問題だと思います。
Linux 版なら動くかもしれません。
nemunemu-nyanko.hatenablog.com
1.インストール
- Arlequin35.zip をダウンロード
- Arlequin35.zip に含まれるすべてのファイルを任意のディレクトリに展開
- メインの実行ファイルである WinArl35.exe をダブルクリックして Arlequin を起動(winArl ってことはwindows 向けなのかな…?mac と違ったらすまない)

2.DNAsp を用いてインプットファイルを作成
わたしはインプットファイルの作成をDNAsp で行っています。
DNAsp のインプットにはfasta ファイルを使います。
ソフトウェアの起動 > File >Open data file >fasta ファイルを選択
上手く読み込めたらData Information という黄色背景のタブが表示されます。
このタブはclose します。
もし、ファイルをArlquin 形式だけではなくNexus 形式でも保存する場合にはデータのフォーマットを設定します。
Data > Format >Nucleotide Sequence Format タブ
ここからがポイントで、Fst のような集団間の比較解析を行いたい場合にはサンプルをグループ分けをする必要があります。
Data > Format >Define Sequence Set...
でDefine Sequence Set タブが開きます。

このList of all sequence のサンプルの中から同じグループにしたいサンプルだけを選択し、真ん中の>> を押すと右側のIncluded List に選択したサンプルだけが移ります。
ご存知とは思いますがShift キーを押しながら選択すると連続選択が、ctrl キーを押しながら選択すると飛ばし飛ばしの選択ができます。何かと使えるので覚えておくと便利です。
同じグループにしたいサンプルを選択 > >> >Add new Sequence Set >グループ名
これを繰り返して全サンプルをグルーピングしたら、Update all entries でグループ情報が書き込まれます。
ちなみに、このときグルーピングした順番でFst が表示されるので、グルーピングの際はお気をつけて。変える方法もあるのかもしれませんが、わたしにはわかりません。
ここまで終わったらファイルを使いたい形式で保存します。
File >Save Export Data as... >Arlequin File Format >OK
設定はお好みにしたらいいと思いますが、Arlequin Haplotype List はつけておきます。
hap ファイルとarp ファイル両方保存します。二つとも同じ名前で同じフォルダに入れます。結果がそのフォルダに吐き出されるので、わかりやすい場所が良いです。
これでインプットファイルの作成はおしまい。
3.Fst の計算
Arlequin を起動します。
Open project >.arp ファイルの選択 >開く
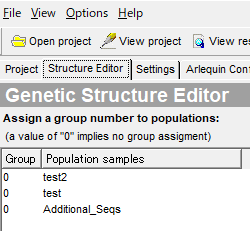
グルーピングが上手くいっていればStructure Editor にグループ分けが表示されます。
Additional Sequence があってわたしもこれが何だか理解できていないのですが、無視して進めて大丈夫です。
Settings >Population comparisons >Compute pairwise FST >distance model 選択
Arlequin で選べるGenetic distance の計算方法は普通のペアワイズディスタンスとReynolds’ distance (Reynolds et al. 1983) とSlatkin’s linearized FST's (Slatkin 1995) があるんだけど、残りの二つについてはわたしも使ったことがありません。おそらく集団サイズや分岐年代を考慮したgenetic distance が出せるんだと思う。詳しくは最初に挙げた公式マニュアルの8.2.4.1 と8.2.4.2 の項目を見てね。
ちなみに残りの二つはhaploidに関するモデルなので、diploid の場合は使いません。
順列(並び替え)検定について
Arlequin では集団間に差がないという仮説(これが帰無仮説にあたる)の下でのペアワイズFST値の帰無分布(帰無仮説を分布化したもの)を計算し、その帰無分布がどのくらいの確率で実測値をカバーするか(p-value)を計算することで有意性検定を行っている(と解釈しています)。
帰無分布についてはこのページがわかりやすいです。
帰無分布(null distribution) | Dr.Clover’s Computer Clinic (fcg.world)
で、この帰無分布を導くための順列計算の回数が「No. of Permutations」、導き出されるp-value を有意とする基準の値を「Significance level」に入れます。
たいていの論文は0.05 にしていますが、それでは大きすぎる、0.01にしろと言う人もいます。分野にもよりけりな感じです。わたしは0.05 のときは数値に「*」、0.01 のときに「**」と印をつけることで弱い有意性と強い有意性を表すことにしています。自分の分野の先行研究を見て決めてください。また、順列計算の回数も先行研究に倣うのが無難です。ただし、大きすぎると計算結果が最後までアウトプットされないエラーが起こるので気を付けましょう。
ちなみにFst の値の解釈に関しては以前記事にしています。参考にどうぞ。
「Fst が高い」の基準のソースってどこ? - Gai memo (hatenablog.com)
あと、Linux 版なら多少の計算の無理は効きます。
4.Mismatch Distribution 解析
データファイルを開くところまではFst と一緒です。
Settings >Estimate parameters of demographic expansion &
Estimate parameters of spatial expansion
これもMolecular distance の出し方が複数あるっぽいのですが、Pairwise distance しか使ったことが無いです。
今回も数値を入れるところがありますが、先ほどと違ってこちらはbootstrap value になります。ザックリ言うと推定パラメータを用いて実行したシュミレーションの回数になります。ブートストラップについては検索してもらえばわかりやすいサイトがいくらでも出てくると思うので説明は飛ばします。
たぶんですけどMismatch Distribution 解析で一番悩むのはモデル選択なんですよね。ただ、わたしもそこに関しては答えを出しあぐねています。前の指導教官に質問してもフィーリングで決めているとしか思えない答えが返ってきたので、みんなどうやって決めているのか逆に気になるくらいです。
マニュアル(p120, 121)からは
- demographic expansion model はシンプルな個体群の集団拡大を想定しているモデル
- Spatial expansion model はある個体群の範囲が最初は非常に狭い地域に限定されていて、その後、時間と空間の経過とともにその範囲が拡大していく場合を想定しているモデル
に読み取れます。しかし、わたしの英語力を信用しないでマニュアルを読んでほしい。
わたしの読みが正しい場合、どんな集団を想定するかによって選べばいいのだと思いますが、正直過去にその生き物がどんな殖え方したかなんて想像に過ぎなくない?と思ったのでわたし自身は次の論文では両方載せようかなと思っています。
最初は、偏差平方和(sum of square deviations (SSD) )、すなわち偏差の合計がより小さい方を選択するつもりだったんですけど、ブートストラップ計算によるSSD 値のp-value が全然0.05 を下回らなくて使わないほうが無難に思いました。その辺はまあお好みで…。
やっと最後です。
5.Neutrality tests
前任者からの引継ぎにこれとFst は同時にできない、と書いてあった気がしたのですが、いまやったら全部同時にできました。もし動かないことがあったら別々にやってみてね。
Settings >Neutrality tests >Infinite site model >Tajima’s D & Fu's Fs
これのNo. of simulated samples はランダムサンプリングを行って解析を繰り返す回数です。これも数千回やれとか16000 回やれとかマニュアルには書いてあるけど、先行研究に倣ったらよいのではないかと個人的には思います。
このブログは引継ぎも兼ねているので一応これらの値の説明を置いておきますね。
Tajima’s D について
Tajima's D - 理学のキーワード - 東京大学 大学院理学系研究科・理学部 (u-tokyo.ac.jp)
この統計量は,πとθの差に基づいているので,自然選択がはたらいていないとD = 0が期待される。したがって,Dが0から有意に異なっていると自然選択がはたらいていると予想される。さらに,遺伝的変異を積極的に維持する自然選択(平衡選択:超優性選択や頻度依存選択)がはたらいているとD > 0となり,有害突然変異を排除しようとする自然選択(純化選択)がはたらいているとD < 0となる。したがって,どのような自然選択がはたらいているか,予想できる。考え方がひじょうに簡単であり,またDNA多型を調べるだけで,Dが計算できるため,広く使われるようになった。Dは本来自然選択の有無を知るための統計量であったが,現在では集団の特性を調べるためにもちいられてもいる。理想集団の要件の1つは集団の個体数が一定であることだが,実際のところ個体数は,環境などの影響により増減する。集団の個体数が増加するとD < 0となり,減少するとD > 0となる。すなわち,Dから過去におきた個体数の増減を推測できる。理想集団のもう1つの要件は,ランダムな交配である。集団構造(集団が複数の分集団に分かれており,分集団間の移住が制限されている状態)があると,(ある個体は同じ分集団に属する個体とは交配しやすいが,別の分集団に属する個体とは交配しにくいので)集団全体ではランダムに交配しているとはいえない。したがって,Dは集団構造の有無を調べるために利用できる。理学系研究科では,分集団化している集団において自然選択がはたらいているといった,Dの値に影響を与える要因が同時に複数ある場合について研究がなされている。
Fu’s Fs について
第6限 Neutralityテスト - ryamadaの遺伝学・遺伝統計学メモ (hatenablog.jp)
- Tajima's test と同様に観測データから得られるThetaPiを用いて、そのThetaの値の下で、集団に認められるべきアレル数と実際に観測されたアレル数との差異をもとにNeutralityを評価する
Fu’s Fs もF負の値であれば、近年の集団拡大を示します。
初心者にもわかりやすそうな日本語の説明が手に入らなくてすみません。
6.蛇足(統計の勉強について)
色々統計ワードが出てきて学部生の人の中には戸惑った人もいたかと思います。
統計、何もわからない…の人におすすめしたい超超々初心者向けテキスト「涙なしの統計学」を紹介して締めます。
ベイズとか最尤推定とかそういう初心者からしたら離れ業みたいな内容じゃなくて、その辺の人からしたら笑っちゃうかもしれない正規分布やt 分布や、その他ノンパラの簡単なやつとかその辺についてがほぼ数式無しで勉強できます。数学苦手な人におすすめ。
わたしは数学がほんっと~~~にできないので、M1 でこれに出会って非常に助けられました。数学ができない理系のみんな、読んでね。
リンクは下に貼ってます。
![[商品価格に関しましては、リンクが作成された時点と現時点で情報が変更されている場合がございます。]](https://hbb.afl.rakuten.co.jp/hgb/224fd45d.d2fb0e8b.224fd45e.beff6ca1/?me_id=1213310&item_id=11018079&pc=https%3A%2F%2Fthumbnail.image.rakuten.co.jp%2F%400_mall%2Fbook%2Fcabinet%2F8838%2F88384035.jpg%3F_ex%3D128x128&s=128x128&t=picttext "[商品価格に関しましては、リンクが作成された時点と現時点で情報が変更されている場合がございます。]")